

Research Highlights
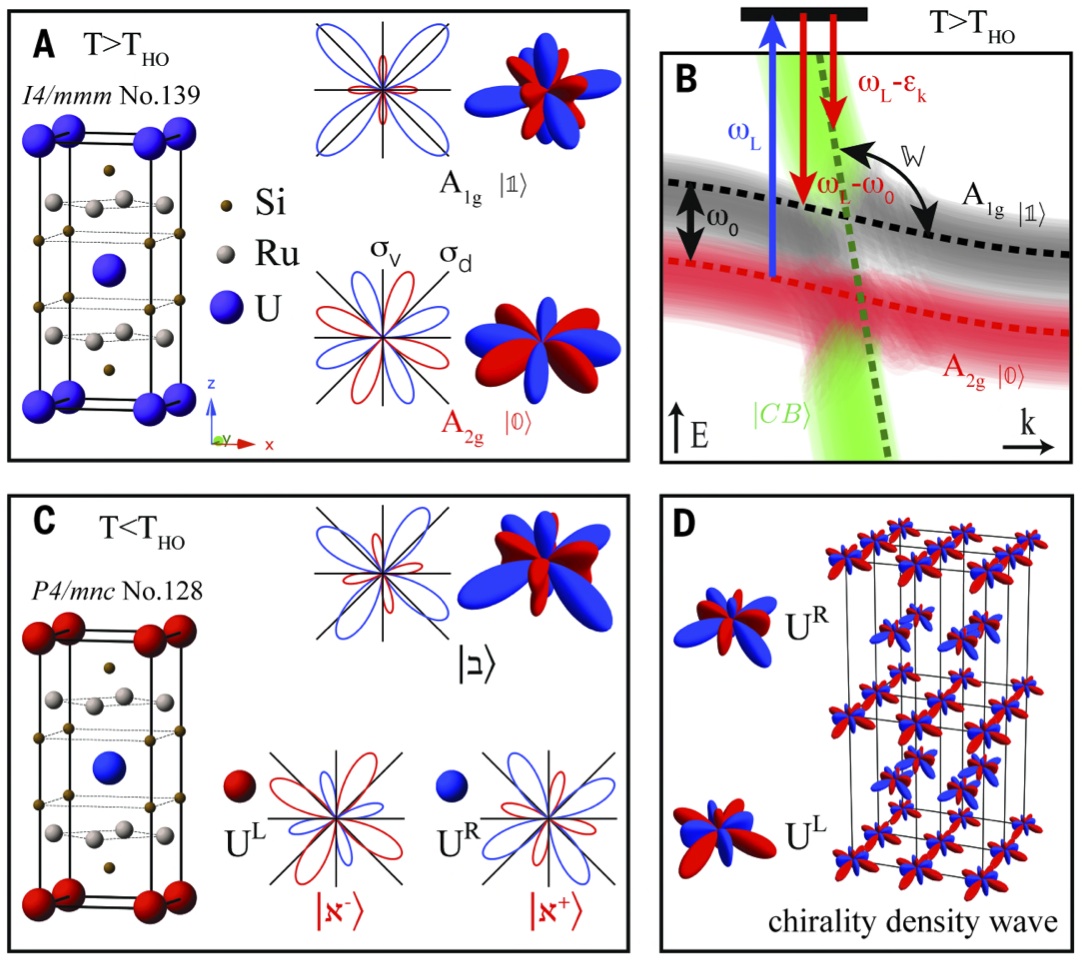
URu2Si2 is a very unique material in which the
complex electronic matter self-organizes at 17.5K in yet unknown
order. A large amount of entropy is lost through the second order
phase transition, a proof that there is an order, but the order
parameter has newer been directly detected in experiment, hence it is
called hidden order.
This "hidden order" has been the subject of nearly a thousand
scientific papers since it was first reported in 1985 at Leiden
University in the Netherlands.
Back in 2009, Gabi Kotliar and Kristjan Haule predicted that the order parameter is a very high order multipole, namely hexadecapole, which is extremely difficult to detect in any experiment.
Girsh Blumberg and his student Sean Kung recently managed to design a very powerful Raman experiment, which undoubtedly showed that the hexadecaple order parameter is broken below 17.5K in this material.
Girsh said: "In this field of correlated electron materials, it is rare to have such predictive power," noting that Gabriel Kotliar and Kristjan Haule developed a computational technique that led to the prediction of the hidden order symmetry.
In the media: Daily Targum, Rutgers Today, ScienceBlog, Materials Today.
Back in 2009, Gabi Kotliar and Kristjan Haule predicted that the order parameter is a very high order multipole, namely hexadecapole, which is extremely difficult to detect in any experiment.
Girsh Blumberg and his student Sean Kung recently managed to design a very powerful Raman experiment, which undoubtedly showed that the hexadecaple order parameter is broken below 17.5K in this material.
Girsh said: "In this field of correlated electron materials, it is rare to have such predictive power," noting that Gabriel Kotliar and Kristjan Haule developed a computational technique that led to the prediction of the hidden order symmetry.
In the media: Daily Targum, Rutgers Today, ScienceBlog, Materials Today.
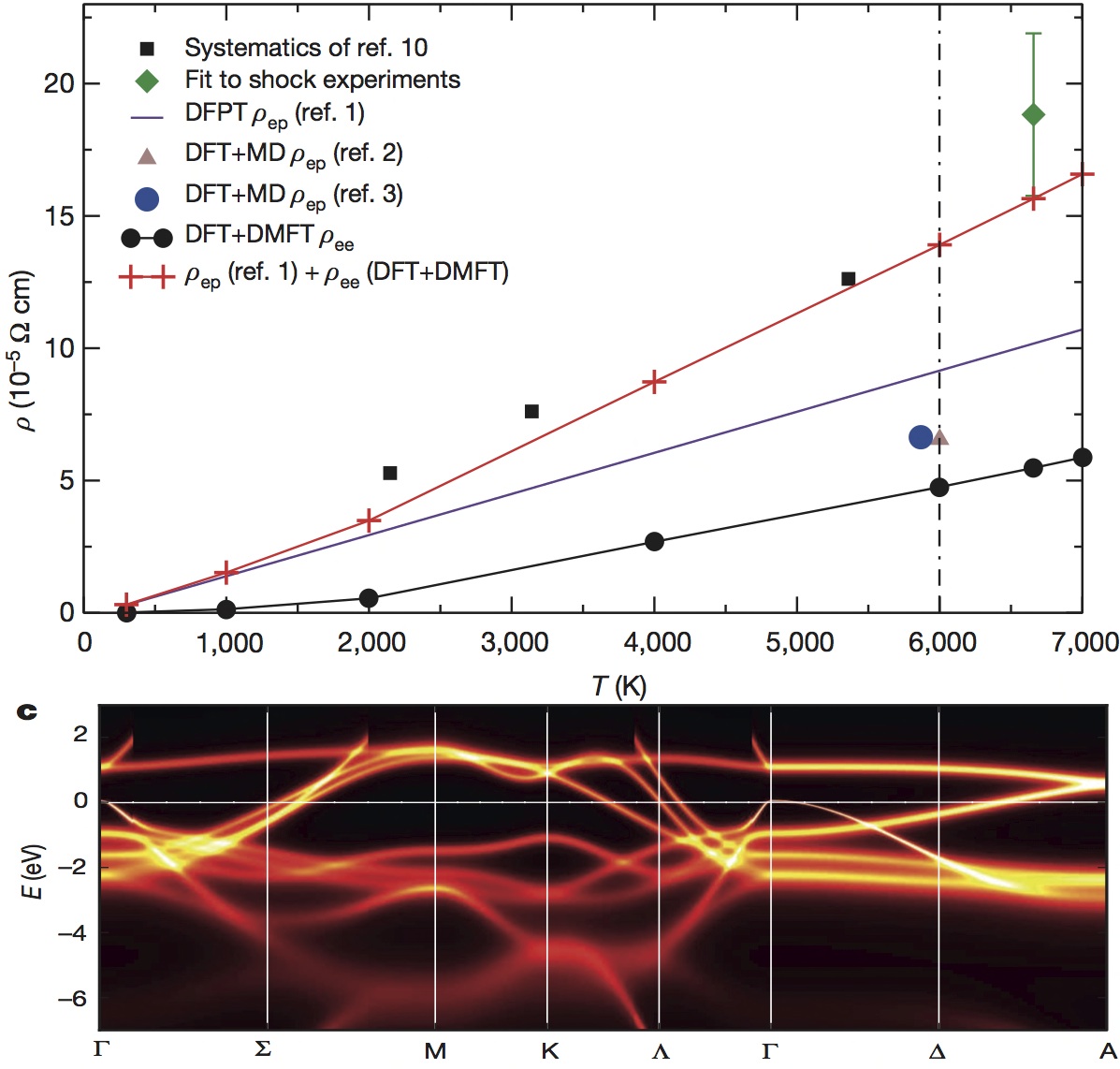
DFT+DMFT just became much more predictive
The electronic structure software developed in our group, which is a combination of Dynamical Mean Field Theory and Density Functional Theory
(DFT+DMFT), has become much more predictive with invention of the exact "double counting" (see Phys. Rev. Lett. 115, 196403). An important problem, which limited the
predictive power of this computational method, was so called double-counting problem. This arrises because both DFT and DMFT account for some part of correlations, but it was not clear unit now what is the common part of correlations accounted by both methods. By the invention of the exact double-counting this ambiguity has been finally set to rest, which makes the predictions of the method much more accurate (for example, the prediction of the total energy of H2 molecule shows only 0.2% error: Phys. Rev. B 91, 155144 (2015).)
The free energy functional of the DFT+DMFT computational method, which is stationary and hence very robust to small numerical inaccuracies, has finally been implemented: Phys. Rev. Lett. 115, 256402 (2015). Up to now people have implemented the non-stationary formula for the total energy, which was not very accurate, and was plagued by errors from implementation details and numerical inaccuracies. While the functional was known for several years, its implementation has not been attempted before, and has only now become clear that the use of this functional is essential for structural predictions.
One examples is the derivation of the force on all atoms and its implementation, which has recently been achieved: arXiv:1602.02819. The calculation of the force using the conventional total energy approach leads to terms which are extremely hard to compute (so called two particle vertex function). By using the stationary free energy functional, no such complicated terms appear, and the computation of the force is very stable and accurate. The prediction of complex crystal structures is now possible.
The free energy functional of the DFT+DMFT computational method, which is stationary and hence very robust to small numerical inaccuracies, has finally been implemented: Phys. Rev. Lett. 115, 256402 (2015). Up to now people have implemented the non-stationary formula for the total energy, which was not very accurate, and was plagued by errors from implementation details and numerical inaccuracies. While the functional was known for several years, its implementation has not been attempted before, and has only now become clear that the use of this functional is essential for structural predictions.
One examples is the derivation of the force on all atoms and its implementation, which has recently been achieved: arXiv:1602.02819. The calculation of the force using the conventional total energy approach leads to terms which are extremely hard to compute (so called two particle vertex function). By using the stationary free energy functional, no such complicated terms appear, and the computation of the force is very stable and accurate. The prediction of complex crystal structures is now possible.
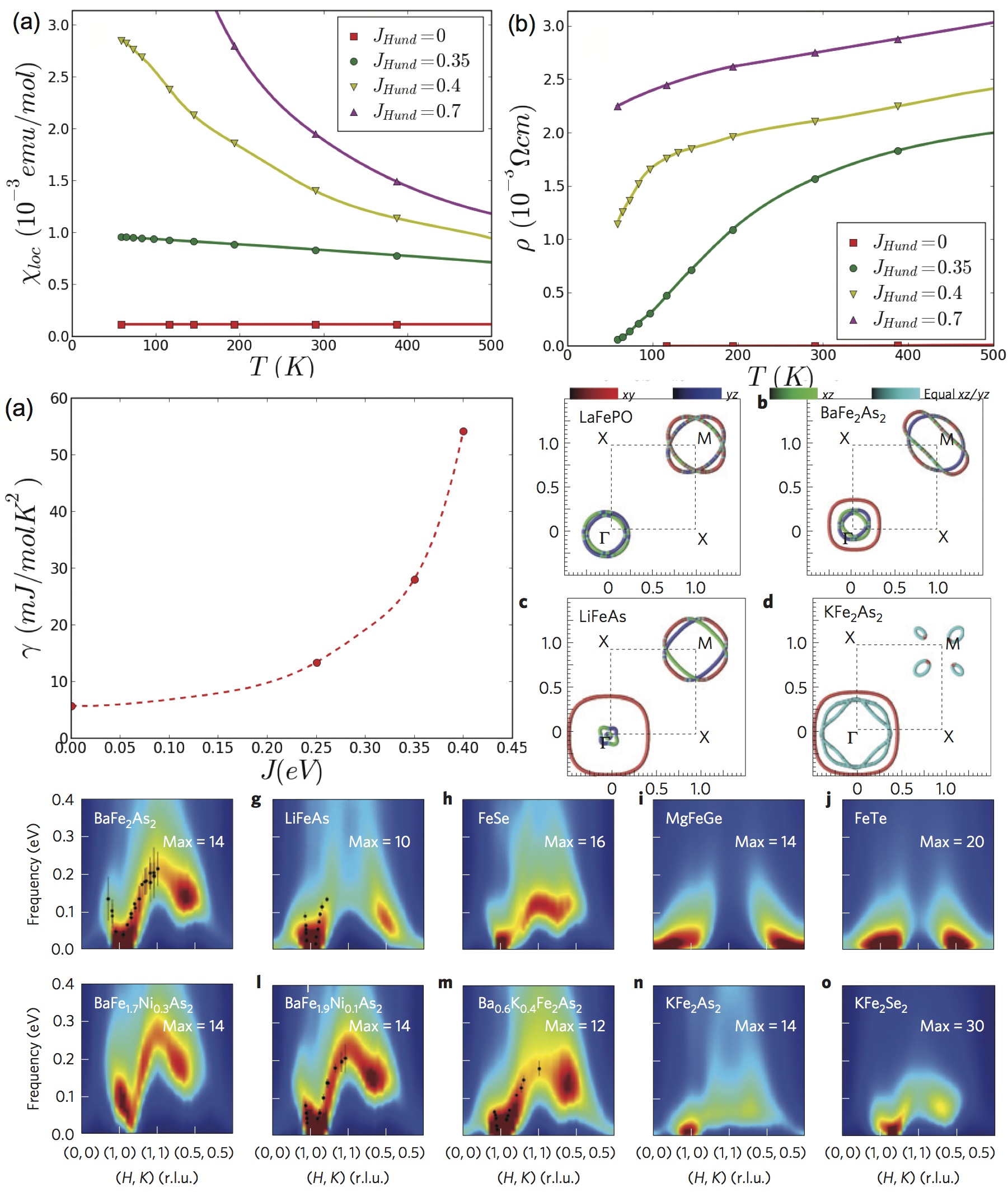
Iron Superconductors are Hunds Metals
Soon after iron superconductors were discovered, we realized that these new superconductors are correlated materials,
hosting unconventional superconductivity (PRL 100, 226402 (2008)),
and we termed them Hund's metals (arXiv 6 May 2008,published in
NJP, see also).
In Hund's metals the Coulomb interaction among the electrons is not strong enough to
fully localize them, but it significantly slows them down, such that low-energy emerging quasiparticles have a substantially enhanced
mass. This enhanced mass emerges not because of the Hubbard interaction U , but because of the Hund’s rule interactions that tend
to align electrons with the same spin but different orbital quantum numbers when they find themselves on the same iron atom.
Our invention of Hund's metals shed new light onto the reach new physics of iron superconductors, as well as strange correlated physics of many other compounds, such as Ruthenades. The All Electron Dynamical Mean Field Theory, which lead us to this understanding, also allowed us to understand many properties of iron superconductors, such as the charge dynamics and static magnetism (Nature Physics 2011), correlation strength and ordered magnetic moments across many families of iron compounds (Nature Materials 2011), spin dynamics and nature of pairing in superconducting state (Nature Physics 2014). Extensive comparison with experiment and remarkable agreement of spin dynamics was documented in Phys. Rev. Lett. 2014 Nature Physics 2012, and Nature Communications 2013.
Our invention of Hund's metals shed new light onto the reach new physics of iron superconductors, as well as strange correlated physics of many other compounds, such as Ruthenades. The All Electron Dynamical Mean Field Theory, which lead us to this understanding, also allowed us to understand many properties of iron superconductors, such as the charge dynamics and static magnetism (Nature Physics 2011), correlation strength and ordered magnetic moments across many families of iron compounds (Nature Materials 2011), spin dynamics and nature of pairing in superconducting state (Nature Physics 2014). Extensive comparison with experiment and remarkable agreement of spin dynamics was documented in Phys. Rev. Lett. 2014 Nature Physics 2012, and Nature Communications 2013.

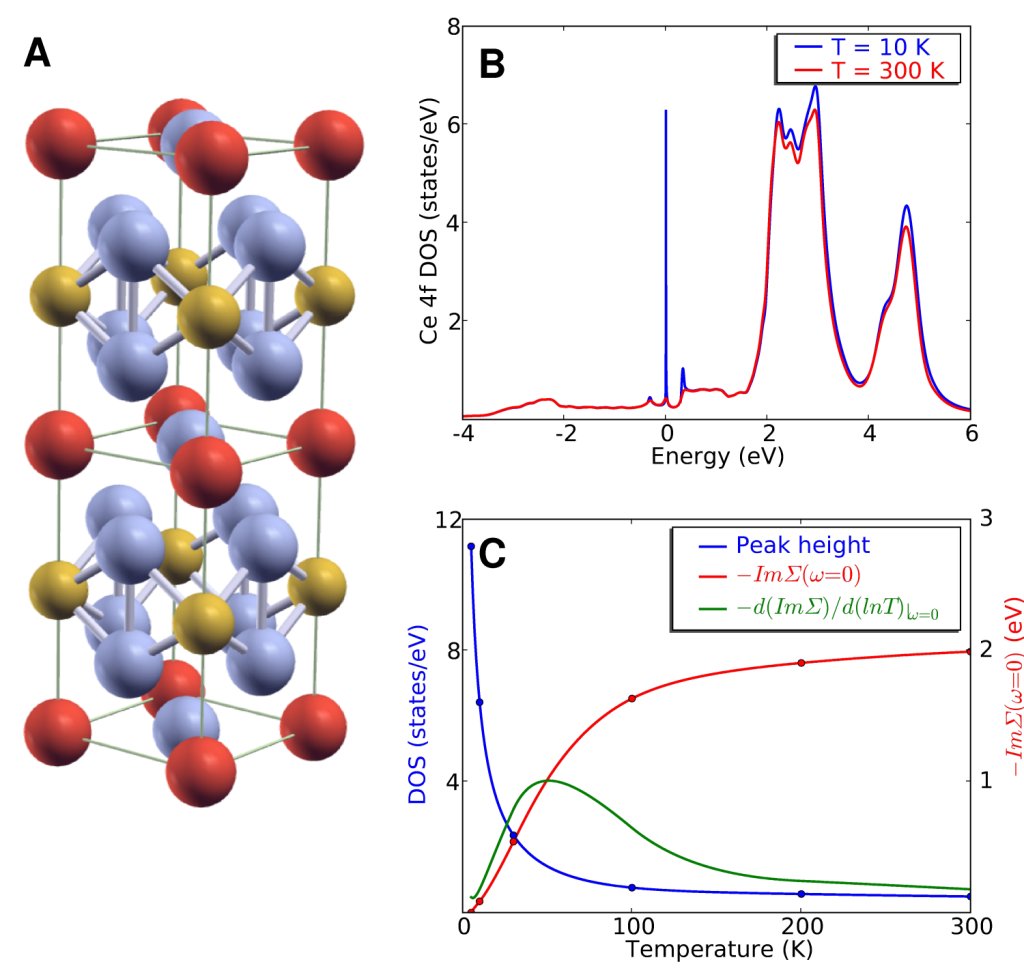
In this Science report we address the
fundamental question of crossover from localized to itinerant state of
a paradigmatic heavy fermion material CeIrIn5. The
temperature evolution of the one electron spectra and the optical
conductivity is predicted from first principles calculation. The
buildup of coherence in the form of a dispersive many body feature is
followed in detail and its effects on the conduction electrons of the
material is revealed. We find multiple hybridization gaps and link
them to the crystal structure of the material. Our theoretical
approach explains the multiple peak structures observed in optical
experiments and the sensitivity of CeIrIn5 to substitutions
of the transition metal element and may provide a microscopic basis
for the more phenomenological descriptions currently used to interpret
experiments in heavy fermion systems.
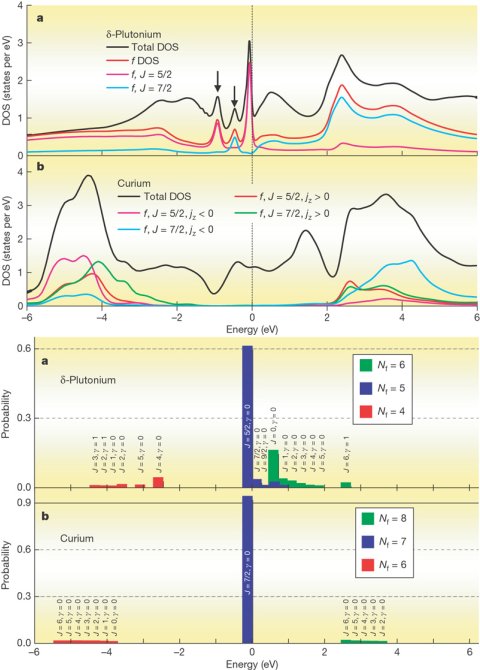
In this Nature letter, we
explain the unique nature of plutonium delta phase, namely its mixed
valence nature, and contrast it to curium metallic phase where f
electrons are localized and order antiferromagneticaly at low
temperature. Curium follows americium in periodic table and is thus
kind of analog of plutonium: plutonium has one hole in americium inert
f-shell (J=0) while curium has one more electron in the americium
inert shell. The striking different properties of the two elements
(one mixed valent non magnetic and other magnetic with Tc=65K) was
hard to understand with any band structure method. We developed
accurate Dynamical Mean-Field Method in combination with LDA and
showed that it describes from first principles the peculiarities of
the two materials. This method is the first that described magnetism
at finite temperature from first principles and show that plutonium is
non-magnetic while curium orders below 100K. This method holds a
great promise that it could predict magnetism from first principles.

Contact
Kristjan Haule, Professor of Physics
Department of Physics and Astronomy
Serin Physics Lab
Office 267
Rutgers University
136 Frelinghuysen Road
Piscataway, NJ 08854-8019
Phone: +1 848-445-9032
email: haule.at.physics.rutgers.edu
Kristjan Haule was born in Slovenia and obtained his
undergraduate education in Slovenia (University of Ljubljana, BSC
1997) and he has done his PhD (2002) work in Slovenia and in Karlsruhe
University (Germany). He was appointed Assistant Professor of Physics
at Rutgers in 2005 and was promoted to Associate Professor in 2009 and
University Professor in 2012.
He was an Alfred P. Sloan Research Fellow in 2008-2010, received NSF Early Career Award in 2008, and The Rutgers Board of Trustees award for Scholarly Excellence in 2009. He is PI on numerous NSF and DOE projects. He received Blavatnik Award for Young Scientists in 2013 for theoretical and computational studies of strongly correlated electron systems.
Haule's research specialties are in condensed matter theory, with major interests in electronic structure theory for correlated electron solids and algorithm development which combine the Dynamical Mean Field Theory and Density Functional Theory. He is especially known for the development of predictive theories for correlated electron solids and implementation of dmft_wien2k code. Haule's publications include over 100 scientific papers, h-index of 35, and over 1,000 citations a year in 2014.
He was an Alfred P. Sloan Research Fellow in 2008-2010, received NSF Early Career Award in 2008, and The Rutgers Board of Trustees award for Scholarly Excellence in 2009. He is PI on numerous NSF and DOE projects. He received Blavatnik Award for Young Scientists in 2013 for theoretical and computational studies of strongly correlated electron systems.
Haule's research specialties are in condensed matter theory, with major interests in electronic structure theory for correlated electron solids and algorithm development which combine the Dynamical Mean Field Theory and Density Functional Theory. He is especially known for the development of predictive theories for correlated electron solids and implementation of dmft_wien2k code. Haule's publications include over 100 scientific papers, h-index of 35, and over 1,000 citations a year in 2014.
Courses taught in the past/present: